Introdução
Epilepsia é uma disfunção cerebral caracterizada pela ocorrência periódica e imprevisível de crises convulsivas, causadas pelo disparo desordenado e rítmico de neurônios, resultando em modificações temporárias do comportamento. Essas alterações ocorrem devido a hiperexcitabilidade e hipersincronismo neuronal, manifestando-se de diversas formas conforme os neurônios envolvidos¹. Em outras palavras, a crise epiléptica é uma descarga elétrica anormal no cérebro que interrompe temporariamente a função cerebral normal, podendo alterar a consciência, causar sensações anormais e movimentos involuntários². As convulsões podem ser precipitadas por: febre, hipoglicemia, hipoxemia, hiponatremia ou hipernatremia, toxinas e trauma craniano³. A epilepsia é definida pela ocorrência de pelo menos duas convulsões, indicando uma predisposição cerebral para crises epilépticas, com crises recorrentes espontâneas separadas por no mínimo 24 horas. Relatos do paciente e de testemunhas são úteis para o diagnóstico, sendo necessários exames complementares, como eletroencefalograma, tomografia de crânio e ressonância magnética, são importantes para a investigação⁴.
Entre os medicamentos mais solicitados para controlar convulsões de diversas origens, destacam-se ácido valproico, carbamazepina, clobazam, etosuximida, felbamato, fenitoína, fenobarbital, gabapentina, lamotrigina, levetiracetam, oxcarbazepina, pregabalina, primidona, tiagabina, topiramato, vigabatrina e zonisamida¹; diazepam, midazolam e lorazepam são os benzodiazepínicos mais utilizados no manejo de crises convulsivas na emergência⁵.
A refratariedade aos medicamentos ocorre quando não se consegue controlar todas as crises epilépticas de um paciente, mesmo após o uso de pelo menos dois medicamentos apropriados. Cerca de um terço dos pacientes apresenta resistência aos medicamentos atuais, dificultando o controle completo das crises⁶, sendo que aproximadamente 1% da população mundial é acometida pela epilepsia¹.
O presente artigo visa não só facilitar a consulta às tipologias de convulsões epilépticas (esquematizadas na sequência), como demonstrar o avanço promissor da cannabis medicinal no tratamento da epilepsia refratária, que vem proporcionando aos pacientes uma redução significativa das crises e uma melhora substancial no bem-estar geral. Além disso, o uso da cannabis por meio da medicina integrativa tem permitido que muitos pacientes reduzam a dosagem de medicamentos antiepilépticos convencionais, os quais frequentemente apresentam efeitos colaterais adversos. Dessa forma, a cannabis vem demonstrando cada vez mais ser uma alternativa terapêutica com menos efeitos negativos e uma forte aliada aos tratamentos convencionais.
Tipos de crises epilépticas convulsivas⁷
TÔNICO-CLÔNICA: Também chamada de “grande mal”, é o que a maioria das pessoas associa ao termo “convulsão”. Elas combinam características de convulsões tônicas (enrijecimento muscular) e clônicas (espasmos rítmicos). Na fase tônica (enrijecimento), todos os músculos enrijecem, a pessoa perde a consciência e cai, podendo emitir um som devido ao ar forçado pelas cordas vocais e, eventualmente, morder a língua ou a bochecha, o que pode causar saliva sangrenta. Na fase clônica (espasmos rítmicos), os braços e pernas sacodem ritmicamente, e a pessoa pode perder o controle da bexiga ou intestino. Após alguns minutos, os espasmos diminuem e a consciência retorna lentamente, podendo haver sonolência, confusão, irritabilidade ou depressão. Essas convulsões duram de 1 a 3 minutos. Se durarem mais de 5 minutos, é necessário atendimento médico imediato, pois isso pode evoluir para status epilepticus, uma condição perigosa que requer tratamento de emergência hospitalar.
AUSÊNCIA ATÍPICA: Também conhecida como convulsões de pequeno mal, são um tipo de convulsão de início generalizado que começa em ambos os lados do cérebro. Durante essas crises, a pessoa pode parecer ausente, mas pode responder um pouco. Movimentos como piscar os olhos, mastigar, estalar os lábios, pequenos movimentos bruscos dos lábios e fricção dos dedos ou mãos podem ocorrer. Detectar esses sintomas pode ser desafiador em pessoas com outros problemas cognitivos ou comportamentais, dificultando a distinção entre uma convulsão e outros comportamentos. Essas crises tendem a começar e terminar gradualmente, ao contrário das crises de ausência típicas que têm início e parada súbitos. Cair durante a convulsão é mais comum nas crises de ausência atípicas, que geralmente duram de 5 a 30 segundos, com a maioria durando mais de 10 segundos.
CONVULSÃO ATÔNICA: O “tom” muscular refere-se à tensão normal do músculo. “Atonica” significa “sem tom”, então, em uma convulsão atônica, os músculos de repente ficam flácidos. Isso pode afetar parte ou todo o corpo, fazendo com que as pálpebras caiam, a cabeça balance ou caia para a frente, e a pessoa pode deixar cair objetos. Se estiver em pé, a pessoa geralmente cai no chão. Essas convulsões geralmente duram menos de 15 segundos, e as quedas podem causar lesões, tornando necessário o uso de proteção para a cabeça, como um capacete. Essas convulsões também são conhecidas como “ataques de queda” ou “convulsões de queda”.
CONVULSÃO MIOCLÔNICA: As convulsões mioclônicas são espasmos breves e semelhantes a choques em um músculo ou grupo de músculos, caracterizados por rápidas contrações e relaxamentos alternados. Normalmente, essas convulsões duram apenas um ou dois segundos, podendo ocorrer isoladamente ou em série em um curto período. Na epilepsia, causam movimentos anormais simultâneos em ambos os lados do corpo e ocorrem em várias síndromes epilépticas:
- Epilepsia mioclônica juvenil: Geralmente envolve pescoço, ombros e braços, ocorrendo frequentemente logo após o despertar. Começa na puberdade ou no início da idade adulta em indivíduos com inteligência normal. A maioria dos casos pode ser bem controlada com medicamentos que devem ser tomados por toda a vida.
- Síndrome de Lennox-Gastaut: Uma síndrome rara que começa na primeira infância e inclui vários tipos de convulsões, incluindo as mioclônicas, que afetam pescoço, ombros, braços e, muitas vezes, o rosto. Essas convulsões são fortes e difíceis de controlar.
- Epilepsia mioclônica progressiva: Envolve uma combinação de crises mioclônicas e tônico-clônicas. O tratamento geralmente é ineficaz a longo prazo, e a condição do paciente tende a piorar com o tempo.
Tabela de Convulsões de Início Generalizado⁸
| Motoras | |
| Crises Tônico-Clônicas | Movimentos rítmicos com enrijecimento. |
| Crises Clônicas | Movimentos bruscos rítmicos sustentados. |
| Crises Tônicas | Enrijecimento generalizado sem movimentos bruscos. |
| Crises Atônicas | Perda do tônus muscular. |
| Início Focal | Crises de Consciência Focais (anteriormente, crises parciais simples) |
| Crises Mioclônicas | Movimentos bruscos rítmicos sem enrijecimento prévio. |
| Crises Mioclônicas-Tônico-Clônicas | Movimentos mioclônicos seguidos de tônicos e clônicos. |
| Crises Mioclônicas-Atônicas | Movimentos mioclônicos seguidos de atonia. |
| Espasmos Epilépticos | Flexão ou extensão súbita dos membros e tronco. |
| Não motoras | |
| Crises de Ausência Típicas | Perda breve de consciência, geralmente sem movimentos. |
| Crises de Ausência Atípicas | Início ou término gradual com alterações do tônus. |
| Convulsões Mioclônicas | Breves movimentos bruscos. |
| Mioclonia Palpebral | Piscar rápido dos olhos. |
Tabela de Convulsões de Início Focal⁸
Com consciência preservada
| Motoras | |
| Automatismos | Movimentos repetitivos sem propósito. |
| Atônica | Perda focal de tônus muscular. |
| Clônica | Movimentos bruscos rítmicos focais. |
| Espasmos Epilépticos | Flexão ou extensão focal dos membros. |
| Hipercinética | Movimentos intensos como pedalar ou debater. |
| Mioclônica | Abalos bruscos focais. |
| Tônica | Rigidez sustentada de um lado do corpo. |
| Não Motoras | |
| Disfunção Autonômica | Sensações gastrointestinais, de calor ou frio, rubor. |
| Parada Comportamental | Parada Comportamental |
| Disfunção Cognitiva | Alterações na linguagem, déjà vu, alucinações. |
| Disfunção Emocional | Ansiedade, medo, alegria. |
| Disfunção Sensorial | Sensações somatossensoriais, olfativas, visuais. |
Com Consciência Comprometida
| Motoras | Mesmos tipos que as motoras com consciência preservada. |
| Não Motoras | Mesmos tipos que as não motoras com consciência preservada. |
Tabela de Convulsões de Início Desconhecido⁸
| Motoras | |
| Crises Tônico-Clônicas | Movimentos rítmicos com enrijecimento de início não claro. |
| Espasmos Epilépticos | Flexão ou extensão súbita com início não claro. |
| Não motoras | |
| Parada Comportamental | Cessação de movimento e resposta sem início claro. |
Medicina integrativa: CBD e qualidade de vida como meta no tratamento da epilepsia⁹
A cannabis e seus extratos orais têm uma longa história de uso no tratamento de distúrbios convulsivos, com registros que remontam há muitos anos e aplicações por neurologistas no final do século XIX. Um exemplo notável é um estudo de 1975 que documentou a recuperação de um paciente com crises não controladas após o início do uso da cannabis¹⁰.
Para uso medicinal, a administração oral é preferida à inalação por permitir melhor controle da dosagem (daí também a preferência pela extração das propriedades). O CBD é extensivamente metabolizado pelas enzimas do citocromo P450, principalmente CYP3A4 e CYP2C19, e apresenta uma eliminação bifásica com uma meia-vida inicial de aproximadamente seis horas e uma meia-vida terminal de cerca de 24 horas devido à sua distribuição lenta nos tecidos.
Além disso, o canabidiol pode aumentar os níveis plasmáticos de outros medicamentos, como o clobazam, potencializando seus efeitos. A combinação com valproato, em particular, tem sido associada a elevações nas enzimas hepáticas. Ainda, o CBD inibe várias enzimas do citocromo P450, o que pode levar a interações clinicamente significativas com outros medicamentos.
Estudos e relatos de casos indicam que a cannabis pode reduzir significativamente as crises em muitos pacientes com epilepsia refratária, como demonstra-se no tópico subsequente. Em Israel¹¹, por exemplo, uma proporção significativa de pacientes apresentou redução nas crises ao usar óleo de cannabis com proporção de CBD/THC de 20:1, com melhorias no comportamento, estado de alerta, linguagem e sono.
Ainda a título de exemplo, outro estudo, realizado nos Estados Unidos¹², com um grande recorte de pacientes com epilepsia resistente a medicamentos, mostrou que o CBD foi bem tolerado e associado a uma redução significativa nas crises em muitos pacientes, especialmente aqueles com síndrome de Dravet e Lennox-Gastaut. Estudos menores e relatos de casos sugerem que o CBD pode ser eficaz no tratamento de convulsões associadas a condições como complexo de esclerose tuberosa, síndrome de epilepsia relacionada à infecção febril, síndrome de Sturge-Weber e crises parciais migratórias malignas na infância.
Estudos de caso
Um estudo publicado no New England Journal of Medicine em maio de 2017¹³ investigou a eficácia do canabidiol (CBD) no tratamento da síndrome de Dravet. Realizado em 23 centros nos EUA e na Europa, o estudo envolveu 120 pacientes (idade média de 9,8 anos) que foram randomizados para receber placebo ou 20 mg/kg/dia de CBD. Durante 14 semanas, incluindo uma fase de titulação de 2 semanas, a frequência média mensal de crises convulsivas caiu de 12,4 para 5,9 no grupo CBD e de 14,9 para 14,1 no grupo placebo.
A redução mediana ajustada na frequência das crises foi de -22,8% no grupo CBD em comparação ao grupo placebo (p = 0,01). No grupo CBD, 43% dos pacientes tiveram uma redução ≥ 50% nas crises, comparado a 27% no grupo placebo. Três pacientes no grupo CBD ficaram livres de crises, contra nenhum no grupo placebo. Este ensaio foi o primeiro a fornecer evidências robustas de que o CBD, quando adicionado ao tratamento existente, reduz a frequência de crises convulsivas em pacientes com síndrome de Dravet.
Outro estudo¹⁴ pertinente foi realizado com pacientes pediátricos com encefalopatias epilépticas refratárias, no Hospital Garrahan {centro pediátrico de referência em saúde pública, gratuito e de alta complexidade}, na Argentina. Foram excluídas crianças sem diagnóstico confirmado, com crises sintomáticas, insuficiência hepática ou renal, alergia ao óleo de CBD ou outras condições de instabilidade clínica. Os pacientes receberam CBD (Rideau®, Aphria, Canadá) em doses iniciais de 2 mg/kg/dia para crianças com peso ≤ 45 kg e 5 mg/kg/dia para as que pesavam > 45 kg, com dose máxima de 25 mg/kg/dia. O produto foi aprovado pelo Health Canada e ANMAT. O estudo acompanhou 59 pacientes entre outubro de 2018 e março de 2020, com idade média de 10,5 anos. A média do tempo de seguimento foi de 20 meses. Os pacientes apresentavam diferentes tipos de encefalopatias epilépticas, sendo a etiologia estrutural (37%), genética (20%), infecciosa (7%), imune (2%) e desconhecida (34%).
No início do estudo, os pacientes tinham uma média de 959 episódios de crise por mês. Durante o estudo, 77,6% dos pacientes tiveram uma redução de pelo menos 25% nas crises, 73,5% uma redução ≥ 50% e 49% uma redução ≥ 75%. A média de crises mensais diminuiu de 959 para 381, uma redução de 60%. Cinco crianças ficaram livres de crises.
Os efeitos adversos mais comuns foram leves a moderados, incluindo sonolência, anorexia, náuseas, vômitos, diarreia, perda de peso, insônia, irritabilidade e mudanças de humor, geralmente resolvidos com ajustes na dose de CBD ou dos anticonvulsivantes, especialmente o clobazam. Os pais relataram melhorias na qualidade de vida dos filhos, incluindo contato visual (81%), motricidade (59%), comunicação não verbal (57%), redução na duração das crises (53%), sorriso social (45%), comportamento e padrão de sono (27%), e comunicação verbal (14%). Os pacientes com síndrome de Lennox-Gastaut (SLG) foram o maior subgrupo no estudo, com 29 dos 38 respondendo bem ao tratamento com CBD, mostrando uma redução nas crises de > 50%.
O último estudo selecionado¹⁵ foi um ensaio clínico de fase 3, multicêntrico, randomizado, duplo-cego e controlado por placebo, realizado em 30 centros (20 nos EUA, 5 na Espanha, 3 no Reino Unido e 2 na França). Os pacientes foram recrutados entre 8 de junho e 15 de dezembro de 2015 e acompanhados por até 24 semanas. O ensaio incluiu um período inicial de 4 semanas, 14 semanas de tratamento (com 2 semanas de escalonamento de dose e 12 semanas de dose estável), um período de redução gradual de até 10 dias e um período de 4 semanas de acompanhamento de segurança após a descontinuação do canabidiol ou placebo. O canabidiol e o placebo foram fornecidos pela GW Pharmaceuticals.
Foram incluídos pacientes com síndrome de Lennox-Gastaut com idades entre 2 e 55 anos, com um eletroencefalograma mostrando padrões característicos e que tinham pelo menos dois tipos de convulsões generalizadas por pelo menos 6 meses. Os pacientes elegíveis estavam tomando entre um e quatro medicamentos antiepilépticos e tiveram pelo menos duas crises convulsivas por semana durante o período inicial. Critérios de exclusão incluíram condições médicas instáveis, histórico de abuso de substâncias, uso recente de cannabis, corticotropinas ou felbamato. Após um período inicial de 4 semanas, os pacientes elegíveis foram randomizados para um dos três grupos de estudo usando um cronograma gerado por computador. Os grupos receberam canabidiol em doses de 20 mg/kg/dia, 10 mg/kg/dia, ou placebo correspondente.
O canabidiol e o placebo foram administrados por via oral duas vezes ao dia. Os pacientes ou cuidadores registraram o número e tipo de convulsões, uso de medicamentos concomitantes e eventos adversos em diários de papel e em um sistema interativo de resposta de voz. As visitas clínicas ocorreram às 2, 4, 8 e 14 semanas após a randomização, com ligações adicionais para avaliar o uso de medicamentos concomitantes e eventos adversos.
Dos 293 pacientes avaliados, 225 foram randomizados: 76 no grupo de canabidiol 20 mg, 73 no grupo de canabidiol 10 mg e 76 no grupo placebo. As características basais foram semelhantes entre os grupos. A maioria dos pacientes era branca e dos EUA, com uma mediana de 6 medicamentos antiepilépticos previamente utilizados e 3 medicamentos concomitantes durante o estudo. A redução média em relação ao valor basal na frequência de crises convulsivas foi de 41,9% no grupo de canabidiol 20 mg, 37,2% no grupo de canabidiol 10 mg e 17,2% no grupo placebo. As diferenças medianas estimadas entre o grupo de canabidiol e o grupo placebo foram significativas (P < 0,01). Percentagens maiores de pacientes no grupo de canabidiol (20 mg: 39%, 10 mg: 36%) tiveram pelo menos uma redução de 50% na frequência de crises convulsivas em comparação com o grupo placebo (14%). A redução percentual na frequência de todas as convulsões também favoreceu o canabidiol.
Eventos adversos foram comuns, com 94% dos pacientes no grupo de canabidiol 20 mg, 84% no grupo de canabidiol 10 mg e 72% no grupo placebo relatando eventos adversos. Os eventos mais comuns incluíram sonolência, diminuição do apetite e diarreia.
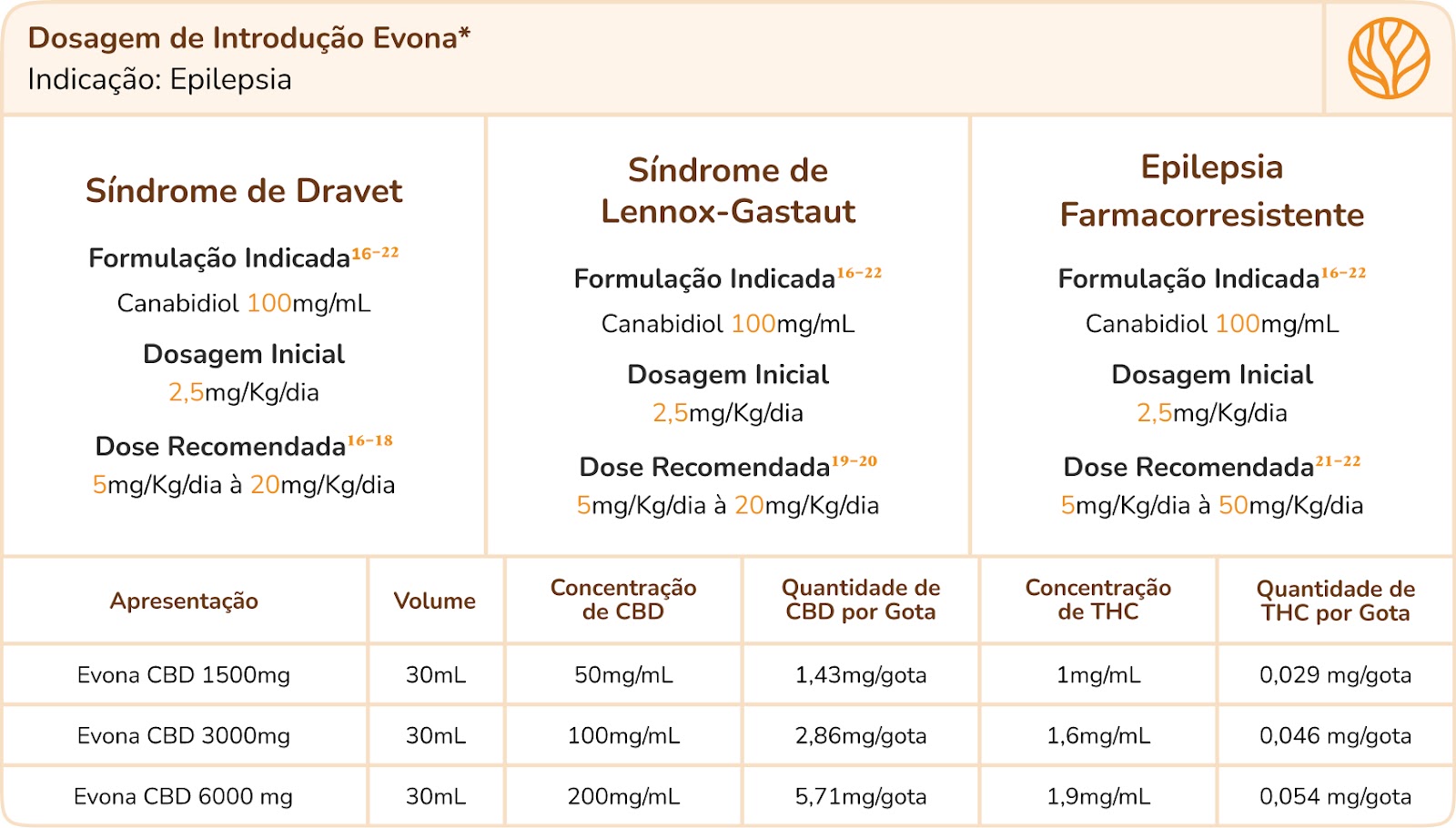
Dosagem
As doses sugeridas não são orientações médicas, apenas uma sugestão de direcionamento com base em estudos científicos publicados sobre o assunto. Para cada indicação clínica estão os artigos científicos para consulta. Na literatura médica, é recomendado alcançar a dose ideal para o paciente através da titulação a partir dos menores valores iniciais e aumentando gradualmente. Em alguns casos, a dose mínima pode ser a melhor dose para o paciente. A recomendação é iniciar com uma dose diária de CBD de 2,5mg/kg (com ou sem THC, geralmente presente em 0,2% nas formulações).
Conclusão
A pesquisa sobre o uso do CBD no tratamento da epilepsia tem mostrado resultados encorajadores. Estudos clínicos e relatos de casos têm demonstrado que o CBD pode reduzir significativamente a frequência das crises em pacientes com epilepsia refratária, especialmente em síndromes severas como a síndrome de Dravet e a síndrome de Lennox-Gastaut. Além de diminuir as crises, o CBD também tem mostrado melhorar a qualidade de vida dos pacientes, contribuindo para melhorias no estado de alerta, humor e padrões de sono.
É importante destacar que o uso do CBD deve ser cuidadosamente monitorado devido às suas interações potenciais com outros medicamentos anticonvulsivantes e aos possíveis efeitos adversos. A dosagem ideal deve ser determinada individualmente, começando com doses baixas (conforme recomendado) e ajustando gradualmente conforme necessário.